
For me, the most enjoyable aspect of discovery research is exploring the unknown. It is about having a big idea; believing in that big idea based on a scientific belief framework; coming to a crossroads in the validity of the big idea, which is usually marked by deep uncertainty and skepticism; making a data-driven scientific decision to proceed (or not) to the next inflection point of testing the big idea; and ultim ately arriving at a conclusion of whether the big idea is true.
Unfortunately, most of these scientific adventure stories are lost in the way we communicate about science. We tell a story to communicate the final message – we have a new medicine that is effective in treating patients – as that is the cleanest way to communicate to an audience not familiar with the gory details of the discovery. Such retrospective narratives are also the simplest way to communicate the validity of the big idea, not the tortuous and often complicated path to arrive at truth.
But such retrospective narratives don’t capture the immensely personal nature of our research discoveries. Moreover, such retrospective narratives often make the big idea seem preordained or obvious, when the big idea was anything but.
In this blog, I tell an important part of the scientific adventure story that led to the discovery a novel medicine called deucravacitinib (Sotyktu™). [Disclaimer: I am a full-time employee at Bristol Myers Squibb. The views expressed here are my own.] In September of 2022, this medicine was approved by the FDA for the treatment of moderate-to-severe plaque psoriasis. While there were many critical moments in the history of deucravacitinib’s approval, I focus on three important – and personal – components in Research, as these discovery moments are often lost in a retrospective narrative.
The discovery of TYK2 dates to 1990 (link here), well before Bristol Myers Squibb (BMS) scientists started working on a drug discovery campaign. It was the second gene fished out of a human B and T cell lymphocyte cDNA library using low-stringency tyrosine kinase probes – hence the name tyrosine kinase 2 (TYK2). It was subsequently determined to be non-membrane bound, signaling downstream of type 1 interferons and IL-23, and structurally related to the Janus kinase (or JAK) family of proteins (here). A definitive link to human disease was established in 2006 with the identification of rare, loss-of-function mutations associated with hyper-IgE syndrome and immunodeficiency (here, here). Between 2007-2012, causal links were established to multiple autoimmune diseases (e.g., Crohn’s disease, systemic lupus erythematosus, multiple sclerosis, rheumatoid arthritis, psoriasis) through genome-wide association studies (GWAS) and related approaches. In 2015, a Phenome-wide Association Study (or PheWAS) was performed demonstrating that the same genetic variants that protect against autoimmune diseases do not lead to increased risk of infection or other side effects associated with JAK inhibitors – the first time any such adverse drug event (ADE) PheWAS was performed (here). And in 2016, a functional genetic study provided more definitive evidence that a protein-coding variant, P1104A, that protects from autoimmunity leads to a 70-80% loss-of-function of TYK2 in human blood cells (here).
But even before these detailed genetic, ADE PheWAS, and functional studies were performed, TYK2 had been identified as an attractive drug target. Accordingly, many pharmaceutical companies, including BMS, were hard at work identifying selective inhibitors of TYK2 in the 2000s. And here is where the BMS story gets personal, tortuous, and exciting!
Discovery moment #1: opening the aperture with phenotypic screens. The problem that BMS scientists faced was the same problem facing others: identifying inhibitors that were sufficiently selective for TYK2 versus the highly related proteins JAK1, JAK2, and JAK3. The reason selectivity is important is that JAK inhibitors are associated with adverse events such as infection, cardiovascular disease, and blood clots. (Over time, the FDA added black box warnings to the label of all JAK inhibitors, although definitive evidence for these adverse events took time to establish.)
The convention at the time was to identify ATP substrate competitive inhibitors against the active, or catalytic site, of a kinase protein. However, there is high amino acid sequence homology among the active sites (JH1 domains) of the three JAK molecules (JAK1, JAK2, JAK3) and TYK2. Accordingly, every time a potent inhibitor of the TYK2 catalytic site was identified, there was always residual inhibition of JAK1-3. BMS scientists considered residual JAK activity to be a No Go for a TYK2 inhibitor (see Figure 1 below, Panel 1).
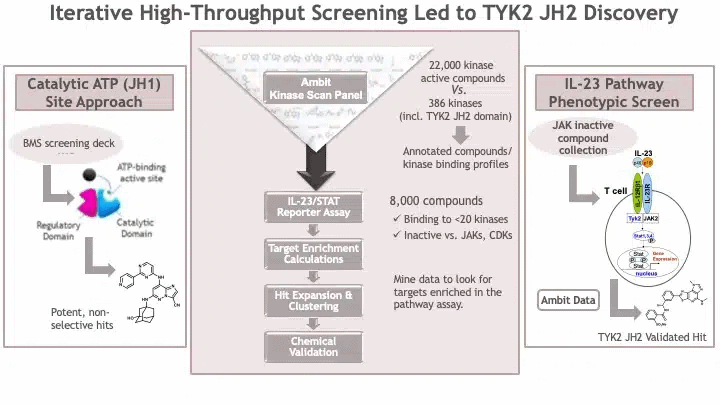
And then, BMS scientists took an alternative approach, the results of which were published by John Tokarski and colleagues in 2015 (here). Rather than focusing solely on TYK2 as the target of interest, they opened the aperture and considered the biological pathway of interest: IL-23 signaling in T cells. The BMS team created a cell-based phenotypic screen assay that could measure IL-23 activity in a cell line. (For a review of phenotypic screens, see here, here.) Specifically, the assay was to measure IL-23 activity in kit225 human T cells with stable integration of a firefly luciferase reporter gene under the control of the interferon-gamma activation sequence. While TYK2 is an important signaling molecule within this pathway, there are others, too – see Figure 1 above (Panel 3). In theory, an inhibitor of IL-23 mediated upregulation of the luciferase reporter could come from inhibiting TYK2, JAK2, STAT1, STAT3, STAT4, or other molecules in the signaling cascade.
A challenge for the IL-23 phenotypic screening assay was that it was difficult to screen many compounds. The convention at the time was to screen millions of compounds using a high-throughput biochemical assay. However, such biochemical assays had to be tailored to a specific aspect of biology (molecular target) – for example, inhibitors of the active site of TYK2. The cell-based IL-23 assay was not able to support testing millions of compounds from the BMS screening deck. Thus, BMS scientists decided to take a multi-step approach.
The first step was to enrich for small molecule compounds likely to be biologically active against TYK2. BMS scientists used a chemogenomics approach, as described in this 2011 publication by Shana Posy and colleagues (Figure 1 above, Panel 2). They applied filters to remove small molecules that were highly promiscuous, active against JAK2 and JAK3, and likely to be cytotoxic to cells. These filters yielded ~8000 small molecules that could be tested in the IL-23 cell-based phenotypic screen.
And BMS scientists held their breaths. Would any of these molecules inhibit TYK2 with the desired characteristics? The results were encouraging albeit perplexing. They identified small molecules that inhibited three targets: GSK- 3-alpha, IKK-beta, and the pseudokinase (JH2) domain of TYK2. The GSK3 result was not surprising, as GSK3a was known to regulate STATs. The IKK-beta finding was initially surprising, as this IKK-beta was not known to be part of the IL-23 signaling cascade. Over time, it was determined that the IKK-beta small molecule also non-specifically inhibited TYK2. What was exciting – and perplexing – was the identification of a TYK2 inhibitor that did not bind to the kinase active site, but to another domain of TYK2 known as the “pseudokinase” domain.
Discovery moment #2: pseudokinase not pseudoscience. The value of an unbiased phenotypic screen is also a potential weakness: identification of small molecules that act through mechanisms that are incompletely understood. For BMS scientists following up on the hits from the IL-23 phenotypic screen, the discovery of a small molecule binder to the pseudokinase domain that had an impact on catalytic activity of TYK2 was at first difficult to explain.
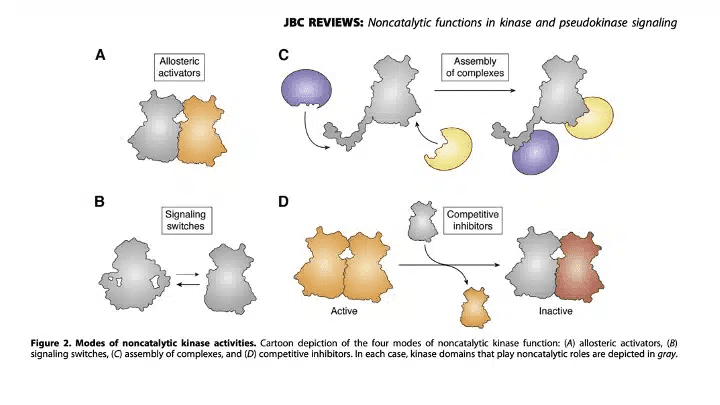
Pseudokinases are defined as pseudoenzymes lacking one or more amino acids required to coordinate ATP and transfer phosphate in catalytically active kinase counterparts (reviewed here). As shown in Figure 2 below, pseudokinases can serve as (a) allosteric regulators of catalytic site; (b) signaling switches; (c) scaffolds to assemble protein complexes; and (d) competitive inhibitors. At the time, most evidence suggested that the TYK2 pseudokinase domain serves as an allosteric regulatory of the catalytic site, although more definitive evidence would not emerge for several more years (here). Making matters more complicated, while dozens of medicines had been developed against catalytic sites, not a single approved drug had been designed to specifically modulate a pseudokinase domain (here). Indeed, deucravacitinib would be the first pseudokinase drug approved by the FDA (here).
Thus, BMS scientists were faced with a challenge: explaining the mechanism by which the binding of a ligand (such as BMS-066) to the pseudokinase domain can prevent receptor-mediated activation of the TYK2 protein by stabilizing the inactive conformation. There were multiple chemical hits such as the imidazo[1,2-b]pyridazine (IZP) compounds described by Ryan Moslin and colleagues in a 2017 publication (here). Resolving the mechanism of these chemical hits required a lot of work, including: (a) biophysical characterization of the binding of hits to the TYK2 pseudokinase domain; (b) cellular characterization of the potency and mechanism of TYK2 pseudokinase domain stabilizers; (c) crystal structure of the TYK2 pseudokinase domain with bound hits; and (d) a model in which a TYK2 JH2/pseudokinase binder could create the specificity of TYK2 inhibition vs that of other JAKs, even though there may be similar biochemical binding to TYK2 vs JAK1 pseudokinase binding.
BMS scientists persevered in the face of uncertainty. They followed the science – not conventional wisdom. They believed in the biology of the target (TYK2) and they believed in the data generated with selective pseudokinase binders. Our chemists continued to pursue novel chemical matter, as described by Chunjian Liu and colleagues in a 2019 publication (here). Together with robust in vivo data generated (which were published in 2019, see here), BMS scientists were increasingly confident that they had discovered a mechanism of TYK2 inhibition that could overcome the JAK selectivity problem plagued by previous efforts from all other companies.
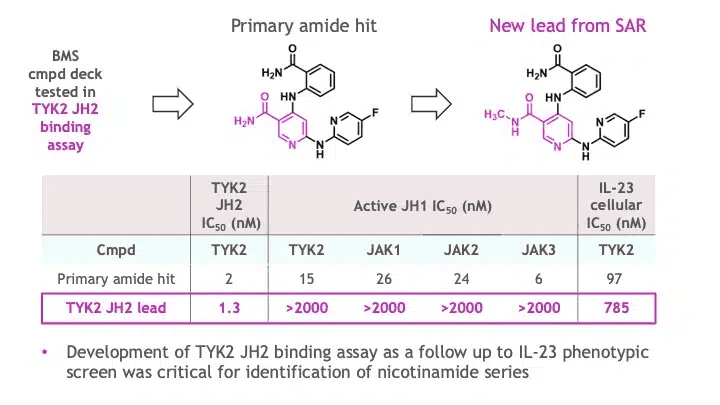
Armed with the hypothesis of JH2/pseudokinase mediated allosteric regulation of the TYK2 catalytic activity, the team conducted a high-throughput campaign with a larger BMS small molecule compound deck to identify direct binders of the TYK2 JH2 domain, as described in a 2019 publication by Ryan Moslin and colleagues (here). The effort delivered a novel chemically diverse nicotinamide hit that, after preliminary structure activity relationship (SAR) assessment, led to a new lead with essentially complete selectivity against JAK JH1 domains as well as excellent general selectivity across the kinome (Figure 3).
But an effective medicine requires a small molecule to have “drug-like properties”, and here is where BMS scientists ran into a third problem that they had to overcome with a novel solution that had not been used before at this stage in drug R&D.
Discovery moment #3: deuterium switcheroo to the rescue. An approved medicine needs to be safe, effective, and sufficiently convenient for patients to take. For an oral medicine to treat common autoimmune diseases such as psoriasis, lupus, and inflammatory bowel disease, this means that convenient dosing (once or twice daily dosing) without liabilities that make taking the medicine complicated for patients (e.g., drug-drug interactions, high pill burden, complex metabolic profile that varies by a patient’s genetic background or with food intake).
A major goal of a late discovery program is to ensure that a medicine that effectively modulates a target of interest (e.g., TYK2) has been optimized for features such as solubility, permeability, metabolic stability, and transporter effects, as these features impact how much drug is absorbed into the blood stream (oral bioavailability), how a medicine is broken down and excreted by the body (metabolism, clearance), and whether a drug has adverse properties (toxicity). These features are commonly referred to as “drug-like properties”.
One TYK2 inhibitor molecule had several desired features – potency, selectivity, and drug-like properties. However, when the molecule was tested in vivo, an unexpected metabolite was observed: one of the small molecule side chains was converted at a high rate into a less selective molecule. BMS scientists incorporated a chemical trick: they changed three of the hydrogen atoms for three deuteriums – a deuterium switcheroo. See Figure 4.
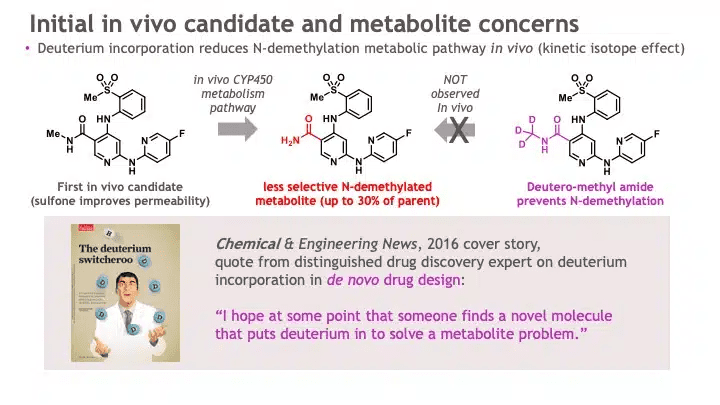
This chemical trick had been in the medicinal chemist’s toolbox for many years, but few drugs had proactively used this heavy-hydrogen trick to effectively solve a problem in lead optimization for a novel mechanism of action, while imparting an overall profile commensurate with a clinical candidate. Most companies use deuteration to breathe new life into older drugs. BMS scientists would become one of the first companies to incorporate deuteration into a new molecular entity. Indeed, the deuterium switcheroo is what gives deucravacitinib the first three letters of its generic name.
There were other optimization steps that led to the final molecule, BMS-986165 (deucravacitinib), including the addition of a cyclopropyl amide to limit a hERG liability, replacement of the pyridine with a pyridazine ring to improve intestinal permeability, and introduction of another heteroaryl group to displace a structural (or high energy) water to improve potency. The first synthesis of deucravacitinib (as described here in a 2019 publication by Stephen Wrobleski and colleagues) allowed for testing in animal models (as described here in a 2019 publication by Jim Burke and colleagues). Ultimately a development candidate was declared, leading to the start of IND toxicology studies and the phase 1 study start at the end of 2015.
In conclusion, the discovery of deucravacitinib is an inspiring example of BMS scientists working courageously and collaboratively on a high-value target, TYK2. In this blog, I describe three discovery moments – pivoting to a phenotypic screen to identify selective inhibitors, unraveling the chemical biology of TYK2’s pseudokinase domain, and incorporating deuterium to avoid a less selective metabolite. There were other important moments, too, as well as many critical contributors (e.g., co-authors on the publications cited in this blog). Such personal discovery adventures are what give me confidence that BMS will continue to be leaders in delivering transformational medicines to patients.
Robert Plenge is a full-time employee at Bristol Myers Squibb. The views expressed here are his own and were originally published here.
About the Author

Robert M. Plenge is Senior Vice President, Research & Development, and Head of Discovery & Translational Sciences (DTS) at Bristol Myers Squibb (BMS). The DTS organization integrates critical functions within Research and Development to deliver on our pipeline of innovative medicines across all therapeutic areas: Informatics and Predictive Sciences, Small Molecule Drug Discovery, Discovery Biotherapeutics, Non-Clinical Research and Development, Clinical Pharmacology & Pharmacometrics, Translational Medicine, and Immunology & Cardiovascular Thematic Research Center. In July 2023, Robert will take over responsibility for all Research activities at BMS as Chief Research Officer. In this role, Robert will be part of the BMS Executive Team, reporting directly to the CEO.
A link to Robert’s full CV can be found here.
This long-form post is the first in a semi-regular series dubbed Profiles in Innovation that feature in-depth scientific content from industry leaders at MassBio member companies. If you are interested in submitting a post for consideration, please contact Ryan Boehm.